 Gli Studi Clinici (Clinical Trials) verificano l’efficacia e la sicurezza, in pazienti, di composti o approccio clinico (preventivo, terapeutico e diagnostico) che hanno il potenziale di modificare in maniera positiva la storia della malattia e quindi la sopravvivenza e la qualità di vita’ del paziente. Tutti i farmaci introdotti nei clinical trials sono stati già ampiamente testati in modelli in vitro della malattia e la loro efficacia e sicurezza investigate in modelli animali, prima che nell’uomo. Solo i trattamenti che mostrano sicurezza ed efficacia massime vengono poi introdotti nei clinical trials.
Gli Studi Clinici (Clinical Trials) verificano l’efficacia e la sicurezza, in pazienti, di composti o approccio clinico (preventivo, terapeutico e diagnostico) che hanno il potenziale di modificare in maniera positiva la storia della malattia e quindi la sopravvivenza e la qualità di vita’ del paziente. Tutti i farmaci introdotti nei clinical trials sono stati già ampiamente testati in modelli in vitro della malattia e la loro efficacia e sicurezza investigate in modelli animali, prima che nell’uomo. Solo i trattamenti che mostrano sicurezza ed efficacia massime vengono poi introdotti nei clinical trials.
Cerca uno Studio Clinico QUI
Studi Clinici: opportunità, arruolamento, approfondimenti
Che opportunità offre partecipare ad un clinical trial?
I farmaci con cui la malattia viene trattata in un clinical trial possono essere dei composti totalmente innovativi non ancora patrimonio della farmacopea nazionale e quindi utilizzati in esclusiva in specifici protocolli terapeutici anche in partnership con altre istituzioni estere aventi le stesse finalità clinico scientifiche; in questo gli Istituti offrono ai nostri pazienti la possibilità di usufruire di approcci terapeutici innovativi e potenzialmente più efficaci ed al tempo stesso tali nuove modalità terapeutiche possano essere istituzionalmente validate per poi divenire patrimonio dell’intero Sistema Sanitaro Nazionale. La tipologia farmaceutica dei prodotti ammessi allo studio può essere rappresentato da molecole sintetiche o biologiche che colpiscono bersagli molecolari noti ma con meccanismi di azione diversi da quelli dei farmaci tradizionali o che colpiscono bersagli molecolari nuovi e noti nel contribuire alla progressione del tumore. Qualunque sia il meccanismo di azione o il target del farmaco sperimentale, esso potrà essere usato come singola terapia o in combinazione con la terapia tradizionale per affiancarla ed aumentarne l’efficacia.
Perchè partecipare ad un clinical trial nei nostri Istituti?
A supporto dei clinical trials i nostri istituti sono in grado di mettere a disposizione avanzate piattaforme per l’inquadramento molecolare della malattia, basate sulla investigazione di molteplici marcatori molecolari. Questo approccio fornisce importanti informazioni diagnostiche e prognostiche , sia all’ inizio del trial che in corso d’opera e la possibiltà di usufrure dellamoderna tecnologia presente e utilizzabile ai fini diagnostici-terapeutici è suffragata da pubblicazioni in riviste dal forte impatto scientifico.
Seguendo tale percorso nei nostri Istituti, diventa attuabile quello che è una delle più recenti acquisizioni concettuali nello studio dei tumori degli ultimi anni: vale a dire la necessità di comprendere, al di là della presenza di uno o più geni malati, il contesto nel quale tali geni difettosi interagiscono e funzionano. Una caratterizzazione dettagliata del contesto in cui le mutazioni del DNA operano (che può essere diverso da paziente a paziente) e’ un fattore estremamente importante che determina la progressione della malattia. Tale tipo di “monitoraggio molecolare” e’ una possibilità reale ed integrata del percorso diagnostico in IFO e perfettamente rispondente alla idea della “personalized medicine” e della ricerca come percorso dal “bench to bedside”.
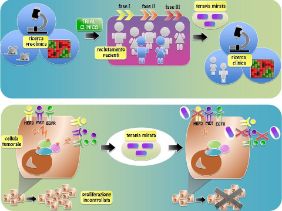
Come funziona l’ arruolamento in un clinical trial?
I clinical trials possono essere promossi dal Ministero della Salute, da Istituti omolohi ai nostri dalle Università o anche da Industrie Farmaceutiche nazionali ed internazionali. In ogni caso, i ricercatori guideranno i trials e seguiranno in dettaglio, con un follow-up giornaliero, l’evoluzione dello studio.
Posso partecipare ad un clinical trial?
L’adesione o meno alla partecipazione ad un clinical trial si può definire come decisione collettiva nel senso che coinvolge in primis il paziente, il suo medico curante, i familiari e, ovviamente, lo staff preposto degli Istituti.
Informazioni essenziali per una decisione tempestiva e corretta sono:
- La diagnosi completa e dettagliata della malattia
- La storia della malattia
Una valutazione approfondita di tali informazioni e’ determinante per stabilire l’ eleggibilità del paziente ad essere arruolato nel trial.
Dal punto di vista del rapporto tra pazienti ed istituzione, verranno fornite con chiarezza le seguenti informazioni, aggiornate al meglio della conoscenza disponibile:
- Sicurezza del trattamento (in termini di effetti collaterali)
- Correnti conoscenze sulla capacità del trattamento di influire sulla storia della malattia
La comparazione con le terapie correnti e sulla attuabilità della terapia sperimentale nel contesto specifico del paziente (genetica, condizioni generali di salute, etc)
- Elementi prognostici personalizzati (quanto tempo libero dalla malattia può garantirmi la terapia corrente?) grazie alla possibilità di inquadrare il paziente a livello molecolare.
Chi guida il clinical trial?
Per ogni clinical trial, viene identificato un direttore dello studio, che sovraintende affinchè lo studio venga svolto nel più rigoroso rispetto delle norme etiche, che la diffusione dei dati (quando disponibili) sia organizzata rigorosamente e tempestivamente e si occupa di mantenere rapporti chiari ed efficaci di collaborazione con tutte le Istituzioni coinvolte nel trial.
I clinical trials non consistono solo nell’utilizzo di molecole a scopo terapeutico.
Sebbene il concetto di clinical trial sia più comunemente associato all’utilizzo di molecole a finalità terapeutica, è importante ricordare che esistono altri tipi di approcci sperimentali che rientrano nella stessa categoria (clinical trials):
- Trials clinici per validare l’efficacia di composti in grado di prevenire l’insorgenza di tumore in soggetti sani (chemoprevenzione) o per prevenire o ridurre la recidiva tumorale in soggetti che hanno già combattuto tale malattia con approcci terapeutici tradizionali.
- Validazione di metodologie diagnostiche innovative per acquisire informazioni più dettagliate sulla stato della malattia, più specifiche possibile per il paziente e quindi in grado di aiutare a fornire approcci terapeutici personalizzati (il tumore è diverso da paziente a paziente, a parità di diagnosi)
- Utilizzo di screening generali di popolazione per acquisire informazioni epidemiologiche sulla distribuzione della malattia e sui fattori di rischio che concorrono alla sua insorgenza e progressione.
- Trials clinici che intendono identificare nuovi approcci per migliorare la qualità di vita dei pazienti sia durante i trattamenti che in ogni altro momento nella storia della malattia.
Fasi del trial clinico
I trials clinici sia in Italia che all’estero, sono composti di varie fasi e la durata di ciascuna fase varia a seconda del tipo di approccio sperimentale che si applica, del tempo di osservazione richiesto per ottenere dati affidabili e della risposta dei pazienti arruolati. Tutto questo, senza dimenticare che lo scopo di ogni trial è quello di capire, in maniera chiara, quanto un nuovo trattamento sia sicuro e quanto sia migliore di quelli attualmente codificati.
Nel caso in cui nel trial si testino nuovi composti a scopo terapeutico, è possibile identificare con maggiore precisione 4 fasi distinte:
- Nella fase 1 i ricercatori testano, in un ristretto gruppo di pazienti, la sicurezza del farmaco, le dosi ottimali di trattamento e operano uno studio dettagliato degli effetti collaterali.
- Nella fase 2 il composto viene somministrato ad un gruppo più numeroso di pazienti per studiarne l’efficacia e definitivamente caratterizzarne gli effetti collaterali.
- Nelle fase 3 l’efficacia del farmaco, da solo o in combinazione con il regime terapeutico tradizionale, viene studiata estensivamente. Si fanno valutazioni comparative con i trattamenti tradizionali e si opera una valutazione definitiva della sua efficacia.
- Nella fase 4, il composto è ormai patrimonio della farmacopea nazionale e come tale in questa fase, la valutazione viene effettuata su gruppi molto ampi di pazienti e l’efficacia e gli effetti collaterali vengono valutati su una scala temporale molto più lunga (anni).
Cosa e’ un placebo e cosa e’ la randomizzazione?
Un placebo consiste in una pillola, liquido o polvere di aspetto identico al farmaco sperimentale testato ma senza effetto. L’uso del placebo è di grande importanza per valutare l’efficacia del farmaco in sperimentazione. Inoltre l’uso del placebo è in genere regolamentato dalla regola del “doppio cieco”, secondo la quale sia il medico somministrante il farmaco sia il paziente che lo riceve non sanno se si tratti del placebo od del composto sperimentale. Quindi, i pazienti verranno organizzati in due gruppi (placebo e trattamento) in maniera tale da non sapere a quale gruppo appartengono.
Come menzionato sopra, i pazienti che sono parte di un clinical trial riceveranno la migliore assistenza possibile, in modo da controllare e valutare immediatamente ogni effetto indesiderato del trattamento possa verificarsi.
Inoltre, il team di medici responsabili del trial può, in caso il trattamento col farmaco sperimentale dia benefici molto superiori rispetto al placebo, decidere di somministrare il farmaco sperimentale anche ai pazienti del gruppo “placebo”, sempre secondo la regola del “doppio cieco”.
In ogni caso, ciascun paziente arruolato ed in qualunque momento, può decidere di abbandonare il trial, previa consultazione con il team responsabile. La decisione del paziente di non continuare con il trial non influirà in alcun modo sulla qualità dell’assistenza da lui ricevuta.
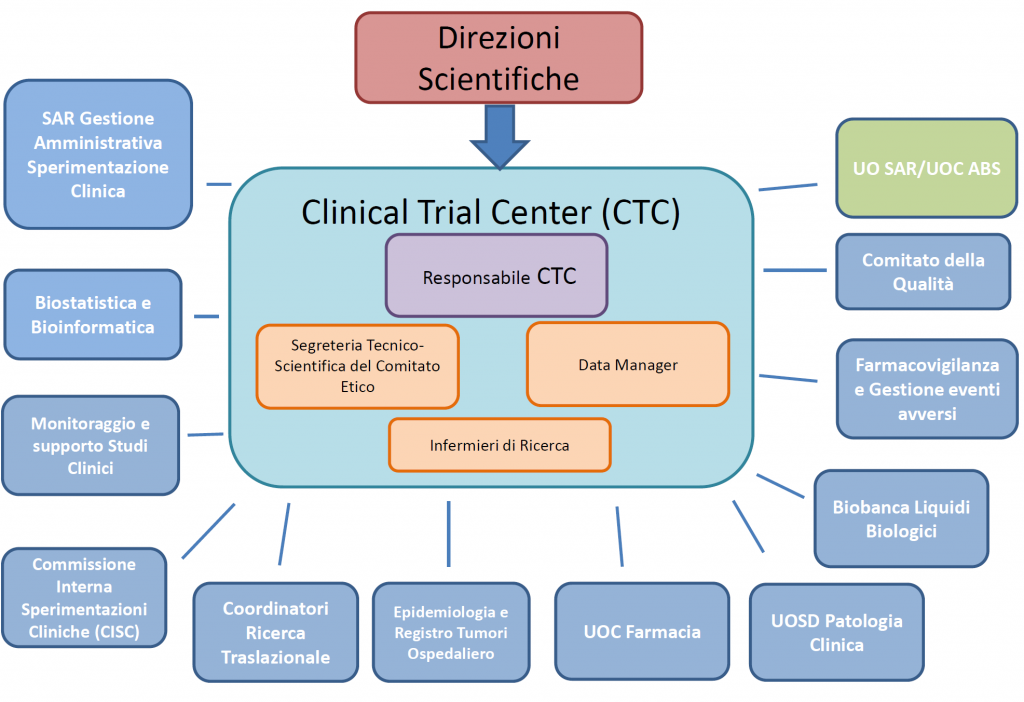
Clinical Trials Center degli IFO
Con delibera n.602 del 6 agosto 2018 gli IFO hanno riorganizzato il proprio Clinical Trial Center.
Il Clinical Trials Center (CTC) è una modalità organizzativa che centralizza e unifica il coordinamento ed il monitoraggio delle sperimentazioni cliniche, con la finalità di fornire servizi di carattere gestionale e metodologico ai ricercatori promuovendo anche la collaborazione tra di loro. Costituisce altresì un fattore incentivante per gli investimenti privati, che prediligono gli Istituti all’interno dei quali è attiva una struttura in grado di sostenere, trasversalmente alle unità operative interne, l’intero processo della sperimentazione clinica (studi di fase II, III, IV). Gli studi di Fase 1 si svolgono presso il Centro Studi di Fase 1 (Early Phase) IFO.
Il CTC svolge le seguenti funzioni:
- coordina e monitora le attività funzionali alla gestione delle sperimentazioni cliniche all’interno degli IFO, ponendosi come punto di riferimento qualificato;
- garantisce alle Direzioni Scientifiche degli IRCCS Regina Elena e San Gallicano ed alla Direzione Sanitaria IFO un maggiore controllo dei processi che riguardano le sperimentazioni;
- supporta in particolar modo la ricerca spontanea no profit;
Si interfaccia con le Unità Operative coinvolte per le attività di ricerca sperimentali coordinandone le attività proprie delle sperimentazioni finalizzate a:
- fornire servizi di carattere amministrativo, gestionale, metodologico e statistico ai ricercatori per l’ideazione, il disegno, la pianificazione, le fasi di start-up, la conduzione, l’analisi e la reportistica di studi clinici affinché queste attività siano eseguite nel rispetto delle Good Clinical Practice (GCP) e dei protocolli stessi;
- supportare la gestione delle procedure autorizzative, la conduzione e la rendicontazione economica di uno studio clinico;
- favorire, nella ricerca profit e no profit, la crescita professionale dei ricercatori coinvolti in termini di rispetto delle GCP e degli aspetti regolatori;
- garantire il controllo della qualità degli studi (sperimentazioni interventistiche e studi osservazionali) con i promotori degli studi profit e no-profit;
- sostenere il monitoraggio delle informazioni riguardo la fattibilità degli studi in termini di pazienti potenzialmente arruolabili;
- incrementare la collaborazione sinergica tra i ricercatori impegnati negli studi;
- valutare le sperimentazioni proposte da ricercatori degli IFO, per le quali gli IFO assumono il ruolo di Promotore, e monitorare lo stato di avanzamento di quelle approvate;
- individuare le aree di maggiore interesse strategico per l’Istituto e proporre iniziative necessarie alla promozione di progetti di sperimentazione clinica in tali aree.
Il personale che lavora presso il CTC deve avere capacità di lavorare in equipe, di relazionarsi con l’esterno, di risolvere criticità emergenti, garantire assenza di conflitto di interessi ed è tenuto al
segreto d’ufficio. Con l’atto deliberativo che recepisce il presente Regolamento di funzionamento del CTC viene contestualmente nominato dal Direttore Generale degli IFO un Responsabile del CTC, su parere dei due Direttori Scientifici e del Direttore Sanitario IFO.
Il CTC dispone, oltre che di personale, anche di uno spazio fisico adeguato che permette nell’insieme di centralizzare la gestione dei data manager (sotto coordinamento del Responsabile del CTC), di ospitare gli infermieri di ricerca, di gestire audits/monitoraggi degli studi clinici con le Ditte Farmaceutiche/Promotori/CRO o con le Agenzie Regolatorie. Inoltre il CTC dispone, attraverso le quote derivanti dalla ripartizione dei fondi provenienti dalle sperimentazioni cliniche, di adeguate risorse finanziarie che possono essere utilizzate anche per il continuo aggiornamento professionale.
Il CTC svolge la sua attività secondo procedure operative standard (SOP) che saranno sistematicamente monitorate tramite audit interni nell’ottica di un miglioramento continuo della
qualità.
Il CTC è composto da:
Responsabile
E’un Dirigente IFO e viene nominato dal Direttore Generale, su proposta dei due Direttori Scientifici e del Direttore Sanitario IFO, sulla scorta dell’analisi curriculare e delle competenze maturate nell’ambito della ricerca clinica applicata Sarà dato rilievo, altresì, alla documentata conoscenza delle GCP ed alla capacità di organizzazione della ricerca e del lavoro di equipe.
Risponde delle attività (inclusi gli obiettivi ed i risultati) del CTC ai due Direttori Scientifici.
La durata dell’incarico è di 3 anni ed è rinnovabile. Il Responsabile del CTC svolge attività di
supporto organizzativo, amministrativo e logistico alle attività di ricerca sperimentali, coordinando tutte le attività funzionali alla gestione delle sperimentazioni cliniche, supportando in particolar modo le iniziative di ricerca spontanea no profit. Garantisce alle Direzioni Scientifiche IRE e ISG il monitoraggio dell’intero processo della sperimentazione clinica, sia di carattere amministrativo che scientifico.
Segreteria Tecnico Scientifica de Comitato Etico
Valida la documentazione, predispone l’ordine del giorno delle sedute del Comitato Etico (CE) e rende disponibile ai componenti il materiale per le sedute stesse. Verbalizza e trasmette i pareri, anche via OSSC (Osservatorio della Sperimentazione Clinica-AIFA). Gestisce il registro delle sperimentazioni cliniche. Gestisce i fondi costituiti dalle quote d’accesso al CE. Gestisce
l’inserimento degli studi e della relativa documentazione nella piattaforma S.M.A.R.T. (Sistema per il Monitoraggio delle Attività scientifico/amministrative della Ricerca Traslazionale). Monitorizza gli eventi avversi in ambito sperimentale con il supporto di personale con adeguata preparazione. Supporta lo sperimentatore nella gestione dei database pubblici (Eudract, clinicaltrial.gov, OSSC).
Coadiuva lo sperimentatore nella gestione degli studi per i quali gli IFO assumono il ruolo di
promotori inclusa l’attività legata all’Osservatorio delle Sperimentazioni Cliniche dell’AIFA e alla sperimentazione sui dispositivi medici.
Data Manager
Collaborano con il medico responsabile, raccolgono la documentazione ed i dati, inseriscono i dati in CRF, gestiscono le queries. Verificano che tutte le procedure siano condotte nel rispetto del protocollo. Gestiscono la contabilità del farmaco. Seguono gli aspetti di notifica degli eventi avversi. Gestiscono le spedizioni ai laboratori centralizzati. Gestiscono e archiviano la documentazione dello studio.
Promuovono la collaborazione tra i diversi reparti nonché con gli altri centri in caso di studi
multicentrici promossi dagli IFO, favorendo lo scambio di informazioni e documenti. Assistono i monitors durante le visite di monitoraggio, gli audit e le ispezioni.
In collaborazione con i responsabili degli studi aggiornano lo stato dello studio e dell’arruolamento sulla piattaforma SMART.
Infermieri di ricerca
Sono coordinati dal responsabile del CTC e selezionati secondo le modalità ed i requisiti di cui alla L. 27 dicembre 2017 n. 205, c.422-434, e successivi decreti attuativi ed al regolamento istituzionale adottato con deliberazione IFO n. 972/2017.
Collaborano con i medici responsabili durante la visita dei pazienti inclusi in progetti di ricerca
monitorando segni e sintomi per identificare possibili eventi avversi, contribuendo a diminuire il rischio per i pazienti e garantendo la qualità nella conduzione dello studio. Assistono direttamente i pazienti rilevando e registrando i parametri vitali ed effettuando i prelievi previsti dallo studio.
Collaborano nella valutazione dei criteri di selezione per l’arruolamento in uno studio. Pianificano visite e prestazioni diagnostiche previste dallo studio. Collaborano con il personale della Biobanca (BBIRE) per la raccolta di campioni come previsto dai protocolli.
Registrano le informazioni sulla somministrazione dei farmaci. Partecipano alle visite di
monitoraggio e agli audit.
Unità che collabora con il CTC:
Gestione amministrativa e sperimentazione clinica
Si occupa della gestione contrattuale ed economica riguardante gli studi. Quando viene proposto uno studio gestisce i rapporti con gli sponsor/promotori, verifica la fattibilità economica dello studio analizzandone costi e ricavi, distinguendo, d’accordo con lo sperimentatore responsabile dello studio, tra attività previste dalla normale pratica clinica e prestazioni specifiche dello studio, definendone quindi il budget e la congruità economica. Negozia i contratti stilati in accordo alla determina regionale e predispone le relative delibere secondo la tempistica prevista dalla normativa vigente. Controlla lo scadenzario dei pagamenti previsto dagli accordi. Gestisce la fatturazione, le entrate e le ripartizioni dei fondi in accordo al budget definito in fase iniziale.
Biostatistica e Bioinformatica
La Biostatistica analizza i dati di tutti gli studi interni agli Istituti, supporta lo sperimentatore nel disegno dello studio e delle schede raccolta dati. Supporta lo sperimentatore nella stesura del protocollo o del progetto di ricerca in accordo con le linee guida prevista per ogni tipologia di studio.
Analizza i dati e ne discute i risultati. Realizza meta-analisi. Crea e sviluppa i database specifici per le attività di ricerca traslazionale e per gli studi retrospettivi. Sviluppa idonee CRF cartacee.
La Bioinformatica crea e sviluppa software web-based necessari per la visibilità degli studi IFO.
Gestisce il database SQL Server che ospita tutto il materiale relativo agli studi clinici. Svolge attività sistemistica del server che ospita i software. Gestisce, in collaborazione con il personale addetto al Monitoraggio e Supporto agli Studi Clinici, gli aspetti operativi e funzionali della piattaforma S.M.A.R.T.
Monitoraggio e supporto agli studi clinici
Questi uffici, distinti per IRE ed ISG e afferenti alle rispettive Direzioni Scientifiche, tramite l’utilizzo della piattaforma S.M.A.R.T., hanno la responsabilità di gestire e coordinare le attività operative legate alla piattaforma S.M.A.R.T. e l’elenco degli studi garantendone la pubblicazione sul sito e mantenendone l’aggiornamento. Supportano inoltre lo sperimentatore nella preparazione della documentazione e, nel caso di studi per i quali gli IFO assumono il ruolo di promotore, gestiscono le pratiche autorizzative, comprese quelle previste dall’attivazione di altri centri, secondo la normativa in accordo con lo sperimentatore responsabile. Gestiscono gli spazi virtuali specificatamente studiati per informare pazienti e medici curanti, in merito alla ricerca clinica condotta nell’Istituto. Elaborano report annuali sulle attività di ricerca clinica. Coadiuvano gli sperimentatori nella presentazione di trial clinici mono e multicentrici interagendo con la segreteria tecnico scientifica del CE. Nell’ambito della rendicontazione annuale della Ricerca Corrente al Ministero della Salute, sono responsabili dell’aggiornamento dei dati relativi ai trial clinici. Gestiscono le procedure di randomizzazione per gli studi promossi dai ricercatori IFO.
Commissione interna sperimentazioni cliniche
Obiettivo principale della CISC è armonizzare l’utilizzo di competenze e risorse dell’Istituto al fine di ottenere un crescente livello di performance nelle sperimentazioni cliniche. In questo contesto ha il compito: a) di valutare le sperimentazioni proposte da ricercatori degli IFO, per le quali gli IFO assumono il ruolo di Promotore, prima della sottomissione al Comitato Etico (CE) (parere vincolante); b) di prendere visione delle sperimentazioni proposte da ricercatori/promotori esterni (parere consultivo). La CISC, poi, monitora lo stato di avanzamento di quelle approvate. La CISC, inoltre, tra le proprie funzioni, ha il compito di individuare le aree di maggiore interesse strategico per l’Istituto e di proporre iniziative necessarie alla promozione di progetti di sperimentazione clinica in tali aree. È composta dai Direttori Scientifici, dal Direttore Sanitario Aziendale, dal Responsabile del Clinical Trial Center, dai Coordinatori della Ricerca Traslazionale, dal segretario del Comitato Etico (senza potere di voto) e dai componenti (IRE /ISG) dell’Ufficio Monitoraggio e Supporto agli Studi Clinici (senza potere di voto).
Coordinatori ricerca traslazionale
I Coordinatori della Ricerca Traslazionale (uno per IRE, uno per ISG) sono responsabili del
monitoraggio degli obiettivi di ricerca traslazionale degli Istituti. Sono membri del CISC e vengono nominati dal Direttore Generale, su proposta dei Direttori Scientifici IRE ed ISG, tra il personale di ricerca a tempo indeterminato degli Istituti. Per la nomina viene dato rilievo al curriculum professionale, con particolare riguardo alla produzione scientifica internazionale, alla capacità di gestione di un gruppo di lavoro nazionale ed internazionale (p.e. responsabile di una UOC o di UOSD o titolare di un incarico professionale di alta specializzazione, ecc) ed alle attività di ricerca traslazionale, come coordinatore oppure come componente di un team di ricerca. Viene richiesto, altresì, l’avere ottenuto in precedenza l’approvazione da un Comitato Etico, in veste di Principal Investigator, di protocolli di ricerca osservazionali o interventistici che prevedano attività di ricerca traslazionale (p.e. utilizzo di materiale clinico da pazienti, ricerca di biomarcatori innovativi, ecc).
Il loro incarico dura 3 anni ed è rinnovabile con nota del Direttore Generale per l’uguale durata. I Coordinatori della Ricerca Traslazionale eseguono un monitoraggio delle attività dei Gruppi di Ricerca Traslazionale (GRIT) di cui si vorranno dotare gli Istituti con atti successivi, e ne rendicontano con cadenza annuale obiettivi e stato di avanzamento dei lavori. Relazionano
periodicamente ai Direttori Scientifici sulle attività dei GRIT, al fine di individuare le strategie più opportune per aumentarne la produttività.
Epidemiologia e Registro Tumori
Questa unità è responsabile del registro tumori ospedaliero, strumento fondamentale per la ricerca clinica. Gestisce il Registro Tumori Ospedaliero. Fornisce informazioni riguardo la fattibilità degli studi in termini di pazienti potenzialmente arruolabili.
Farmacia
È responsabile della gestione dei farmaci sperimentali dalla ricezione della delibera autorizzativa alla preparazione/consegna del farmaco. Gestisce gli studi in cieco e doppio cieco. È responsabile del mantenimento del farmaco alle temperature previste. Svolge attività di formazione a favore del personale infermieristico per la preparazione dei farmaci. Gestisce i farmaci ad uso terapeutico autorizzati dal Comitato Etico. Segue le procedure di smaltimento del farmaco previste per ogni singolo studio.
Qualora la sperimentazione preveda l’impiego di dispositivi medici (DM), il farmacista assicura la buona tenuta del DM, classifica il DM nella classe di appartenenza, forma gli utilizzatori sul corretto utilizzo, collabora all’analisi del rischio legato all’utilizzo del DM, archivia la documentazione amministrativa.
Patologia Clinica IRE
La Patologia Clinica IRE supporta lo Sperimentatore eseguendo gli esami ematochimici
richiesti dallo Studio e al contempo provvede alla gestione dei campioni biologici provenienti dalle attività delle Sperimentazioni Cliniche al fine di centralizzare e garantire gli aspetti qualitativi di tutti i prelievi che vengono eseguiti e che devono essere inviati al centro di raccolta. L’accettazione dei campioni avviene tramite il sistema informatico del Laboratorio (LIMS) che garantisce la tracciabilità dei dati analitici e dei referti diagnostici.
Il personale promuove la corretta gestione delle tecnologie strumentali presenti in Laboratorio,
verifica il piano operativo delle indagini richieste per i pazienti arruolati nello studio verificando la congruità dei campioni sia per le finalità diagnostiche sia per le esigenze di trattamento e conservazione dei campioni fino alla spedizione al Laboratorio di riferimento degli studi clinici.
Biobanca Liquidi Biologici (BBIRE-LB),
La BBIRE-LB nell’ambito delle sperimentazioni cliniche offre ai pazienti che abbiano dato il loro consenso un servizio di raccolta, conservazione e distribuzione del materiale biologico per finalità di ricerca, nel rispetto delle norme etico–giuridiche, e con i più elevati standard tecnologici. Il livello di anonimità dei prelievi e dei dati personali è garantito da un codice con il quale vengono contrassegnati i campioni sotto la responsabilità della Direzione Scientifica dell’IRE; soltanto i Soggetti Autorizzati possono collegare questo codice al nominativo del paziente donatore.
La BBIRE-LB si avvale di un sistema di monitoraggio della temperatura dei congelatori, gestito da un software che riceve e registra i parametri in tempo reale, con diverse tipologie di allarme e di un sistema di gestione campioni in grado di salvaguardare la privacy dei pazienti, registrare i dati relativi ai campioni, inclusa la collocazione fisica e l’identificazione univoca, ed associarli ai dati clinici.
Farmacovigilanza e Gestione eventi avversi
È responsabile dell’attività di farmacovigilanza. Gestisce, in accordo alla normativa, gli eventi avversi per gli studi con promotore IFO (e/o, nel caso di promotore esterno, per i soli studi osservazionali), segnalando gli stessi nelle reti e sistemi di monitoraggio e vigilanza previsti, predisponendo un report trimestrale o semestrale degli eventi avversi osservati presso gli IFO al fine di caratterizzarne le cause e i possibili interventi preventivi, effettuando visite periodiche nei reparti per valutazione delle ADR con medici, infermieri e tecnici, informando sulla sicurezza dei farmaci ed inviando agli operatori sanitari note informative AIFA, comunicati EMA e articoli di letteratura.
Comitato della Qualità IFO
Verifica che gli studi mantengano lo standard di qualità. Esegue audit interni di sistema definiti come revisione continua della qualità ed audit mirati su studi specifici. Supporta gli sperimentatori nel condurre gli studi secondo GCP eseguendo il monitoraggio degli studi interni o per i quali l’istituto è promotore e controlli di qualità sull’andamento degli studi industriali e non.
SAR/ABS
A seguito della contabilizzazione dei proventi delle sperimentazioni cliniche da parte della Segreteria del CE e dell’emissione fattura da parte della UOC Risorse Economiche, la UO SAR genera un codice.
Contatti
Per richiedere maggiori informazioni sugli studi clinici o sul reclutamento dei pazienti scrivere al seguente indirizzo email: